
Genomics Specialist
Contact Krista
Uncovering the taxonomic origin and functional profile of gene sequences is of high interest to biosecurity professionals and fundamental researchers, but it is a challenging and nuanced task to navigate. SeqScreen™, an open source bioinformatics workflow, leverages multiple open source tools to predict the most probable source organism, the diversity of possible taxonomic lineages, functional gene annotations, and customized pathogenicity labels for a given query sequence. SeqScreen™ is useful for annotating full-length gene sequences in assembled genomes, as well as gaining the most information possible from short gene fragments in metagenomes, metatranscriptomes, or individual sequences (>50bp). The SeqScreen™ workflow continues with S2Fast, a tool which fills a critical capability gap in identifying biothreats based on their predicted functional potential to do harm. S2Fast enables the detection and characterization of novel or synthetically designed biothreats, which can more easily evade current state-of-the-art detection methods because they may not resemble previously observed pathogens.
SeqScreen+S2FAST workflow development was conducted by a team of collaborators, including Signature Science and Dr. Todd Treangen’s Laboratory at Rice University, and funded through IARPA’s Fun GCAT program.
SeqScreen + S2Fast Workflows
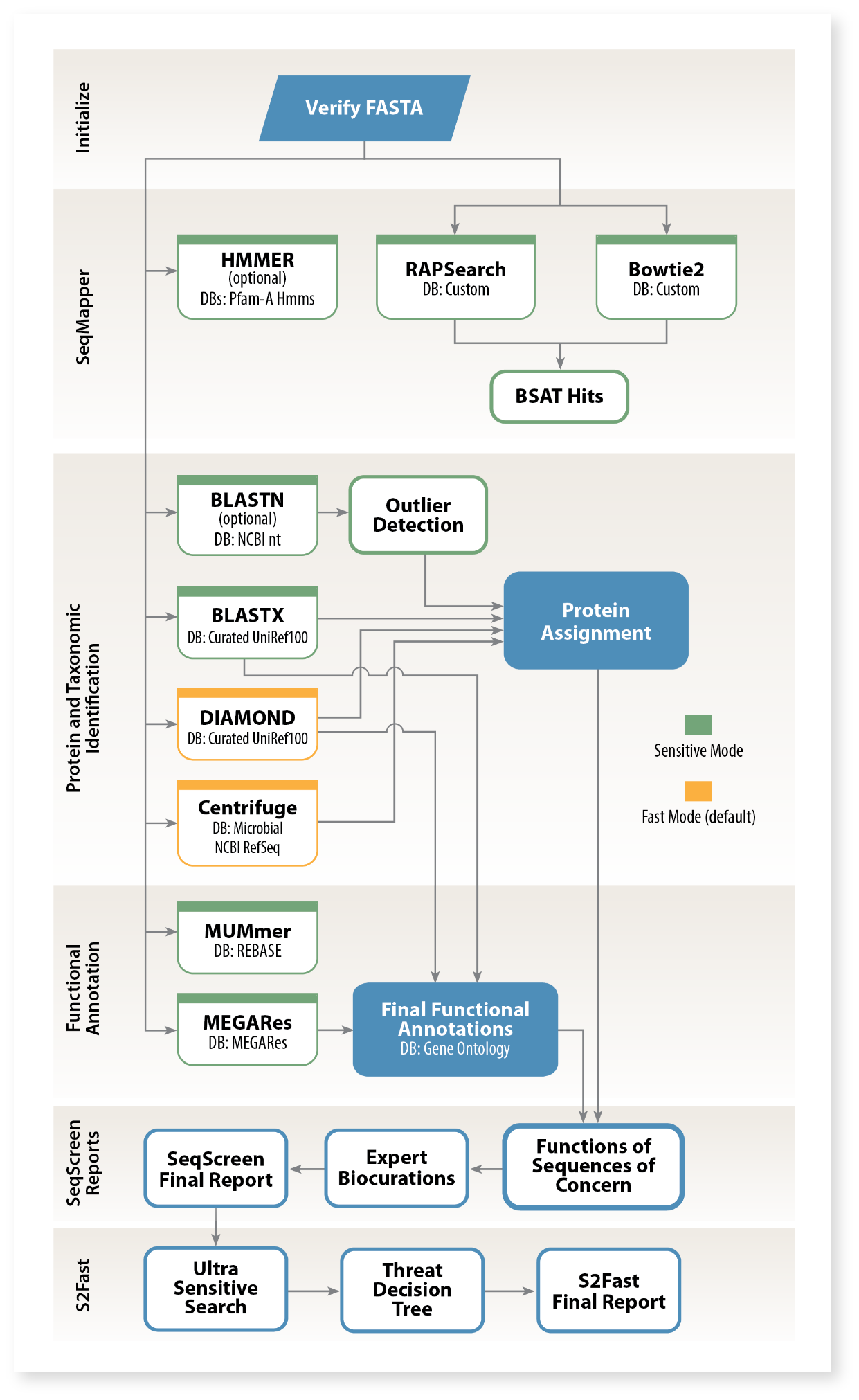
S2Fast predicts the functional threat potential of short sequences from gene fragments. SeqScreen™ first assigns functions using controlled vocabularies, including Gene Ontology (GO) terms, UniProt IDs, and a custom Functions of Sequences of Concern (FunSoCs) schema. Because GO terms were not designed to solely illustrate mechanisms of pathogenicity, FunSoCs were devised to describe the cellular processes relating to the virulence potential of short protein-coding sequences. Signature Science’s microbial virulence subject matter experts (SMEs) reviewed thousands of scientific papers to concisely describe how sequences of concern affect the host deleteriously, and these annotations were expanded to additional sequences with an ensemble machine learning approach. S2Fast leverages a protein database of millions of SME-curated and machine learning-assigned FunSoCs to predict the pathogenic potential of each query sequence and provide a relative threat level (i.e., high threat, moderate threat, mild threat, or negligible threat).
With the SeqScreen+S2Fast workflow, the biothreat community can move beyond matching sequences to short lists of known biothreats toward predicting the threat potential of sequences based on their predicted functions.
Publications
Want more information about S2Fast and SeqScreen™?