
Genomics Specialist
Contact Krista
A bioinformatician’s typical metagenomic data processing roadmap may include goals of producing high-quality Illumina sequence reads with adapters removed, reads assembled into longer contigs, taxonomic content identified within each sample, analysis of functional elements, and interpretation and comparison of results. They will spend a lot of time individually researching, installing, and running the many metagenomics analysis tools available to reach these goals. MetScale™, an open source bioinformatics workflow developed by Signature Science and its academic partners, can expedite and unify this process.
MetScale™ is a suite of automated workflows that reproducibly runs open source metagenomics tools and collectively analyzes the results. MetScale™ enables more accurate interpretation of results by analyzing taxon assignments that are detected across multiple tools and includes a database query tool that determines if a false positive call was the result of an organism not being present in the reference database.
Metagenomic Toolbox – Typical Processing Roadmap
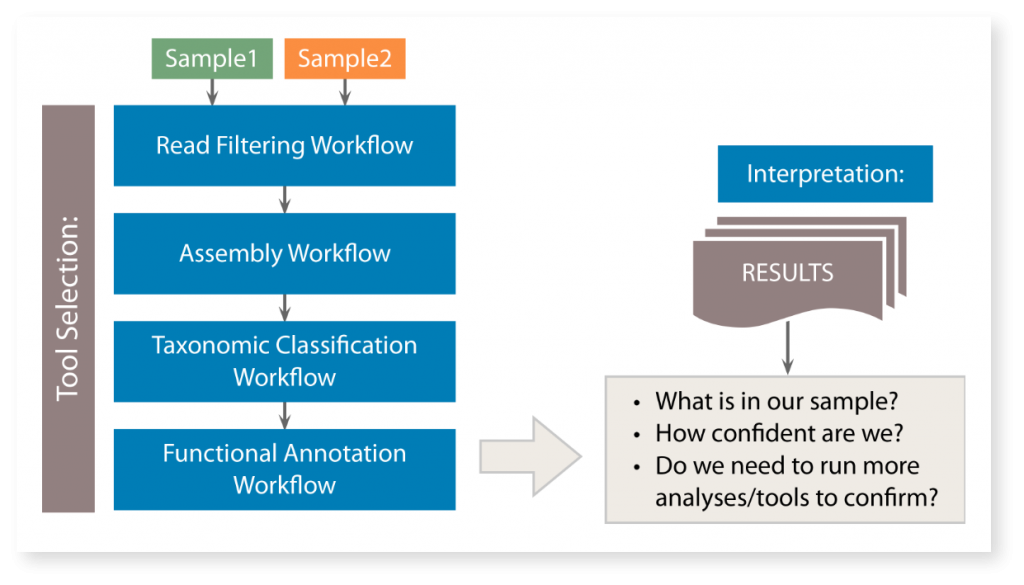
Overview of MetScale™ Workflows
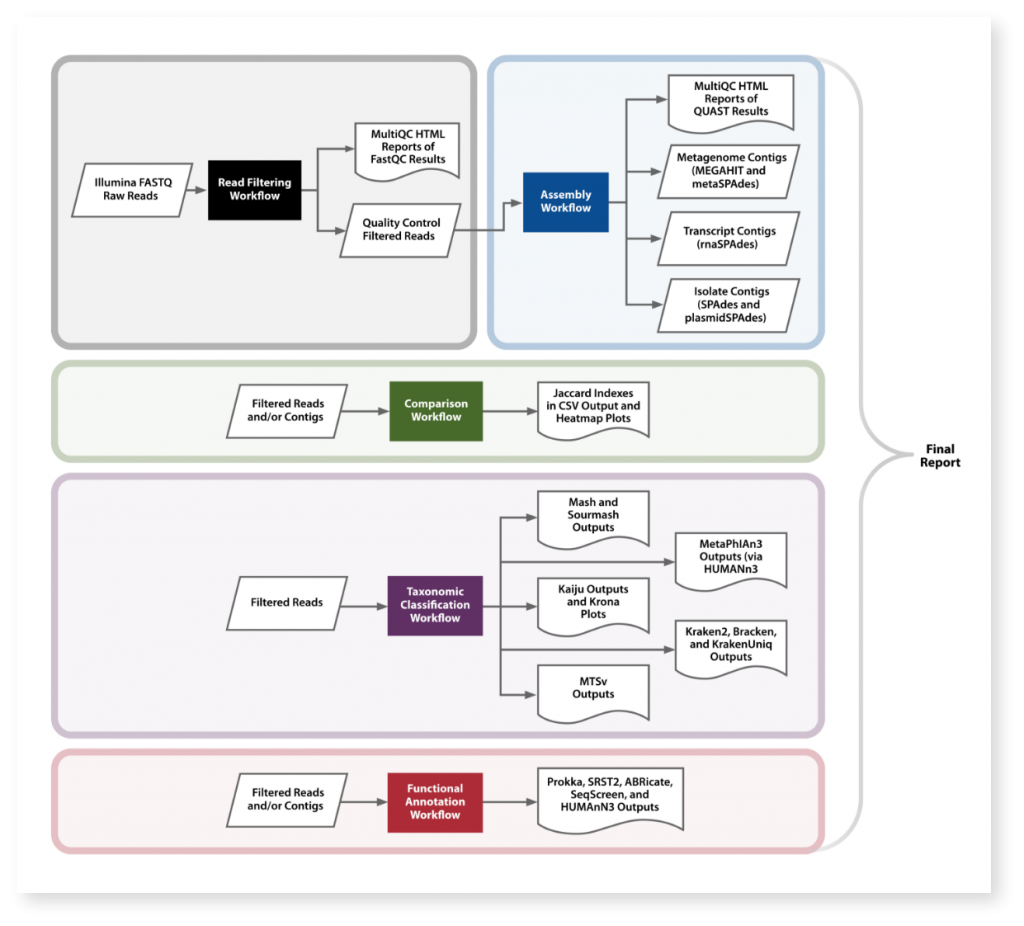
MetScale™ was developed by team members from Signature Science, University of California Davis, Rice University, University of Virginia, and the University of North Texas. Tools used within MetScale™ were developed by many different members of the open source bioinformatics community.
Want more information about Metscale?